Your new post is loading...
Your new post is loading...
Tiragolumab, an anti-TIGIT antibody with an active IgG1κ Fc, demonstrated improved outcomes in the phase 2 CITYSCAPE trial (ClinicalTrials.gov: NCT03563716 ) when combined with atezolizumab (anti-PD-L1) versus atezolizumab alone1. However, there remains little consensus on the mechanism(s) of response with this combination2. Here we find that a high baseline of intratumoural macrophages and regulatory T cells is associated with better outcomes in patients treated with atezolizumab plus tiragolumab but not with atezolizumab alone. Serum sample analysis revealed that macrophage activation is associated with a clinical benefit in patients who received the combination treatment. In mouse tumour models, tiragolumab surrogate antibodies inflamed tumour-associated macrophages, monocytes and dendritic cells through Fcγ receptors (FcγR), in turn driving anti-tumour CD8+ T cells from an exhausted effector-like state to a more memory-like state. These results reveal a mechanism of action through which TIGIT checkpoint inhibitors can remodel immunosuppressive tumour microenvironments, and suggest that FcγR engagement is an important consideration in anti-TIGIT antibody development. A high baseline of intratumoural macrophages and regulatory T cells is associated with better outcomes in patients with non-small cell lung cancer treated with atezolizumab plus tiragolumab, but not with atezolizumab alone.
The CAR T-cell pioneer developed breakthrough therapies for blood cancer patients. Now he has turned his attention to solid-tumor cancers.
Viruses and virally derived particles have the intrinsic capacity to deliver molecules to cells, but the difficulty of readily altering cell-type selectivity has hindered their use for therapeutic delivery. Here, we show that cell surface marker recognition by antibody fragments displayed on membrane-derived particles encapsulating CRISPR–Cas9 protein and guide RNA can deliver genome editing tools to specific cells. Compared to conventional vectors like adeno-associated virus that rely on evolved capsid tropisms to deliver virally encoded cargo, these Cas9-packaging enveloped delivery vehicles (Cas9-EDVs) leverage predictable antibody–antigen interactions to transiently deliver genome editing machinery selectively to cells of interest. Antibody-targeted Cas9-EDVs preferentially confer genome editing in cognate target cells over bystander cells in mixed populations, both ex vivo and in vivo. By using multiplexed targeting molecules to direct delivery to human T cells, Cas9-EDVs enable the generation of genome-edited chimeric antigen receptor T cells in humanized mice, establishing a programmable delivery modality with the potential for widespread therapeutic utility. Cell-specific molecular delivery with enveloped delivery vehicles enables genome editing ex vivo and in vivo.
The success of chimeric antigen receptor (CAR) T cell therapy in treating several hematopoietic malignancies has been difficult to replicate in solid tumors, in part because of T cell exhaustion and eventually dysfunction. To counter T cell dysfunction in the tumor microenvironment, we metabolically armored CAR T cells by engineering them to secrete interleukin-10 (IL-10). We show that IL-10 CAR T cells preserve intact mitochondrial structure and function in the tumor microenvironment and increase oxidative phosphorylation in a mitochondrial pyruvate carrier-dependent manner. IL-10 secretion promoted proliferation and effector function of CAR T cells, leading to complete regression of established solid tumors and metastatic cancers across several cancer types in syngeneic and xenograft mouse models, including colon cancer, breast cancer, melanoma and pancreatic cancer. IL-10 CAR T cells also induced stem cell-like memory responses in lymphoid organs that imparted durable protection against tumor rechallenge. Our results establish a generalizable approach to counter CAR T cell dysfunction through metabolic armoring, leading to solid tumor eradication and long-lasting immune protection. CAR T cells engineered to express IL-10 eradicate solid tumors and metastases.
Identification of ATXN3 as a PD-L1–positive regulator through unbiased CRISPR screening. To identify the specific deubiquitinases that promote tumoral PD-L1 expression, we first designed a targeted library of all 96 mammalian deubiquitinase family members based on the optimized single-guide RNA...
Sialic acid-binding immunoglobulin-like lectin 15 (Siglec-15) is an immune checkpoint molecule with sequence homology to programmed cell death ligand 1 (PD-L1), which is mainly expressed on macrophages and tumor cells. However, whether Siglec-15-induced immunosuppression and poor prognosis are independent of PD-L1 remains unclear. In this study, we collected samples of 135 non-small cell lung cancers and found that Siglec-15 and PD-L1 expression were independent in non-small cell lung cancer by multiple immunofluorescence staining. Siglec-15 on macrophages (Mφ-Siglec-15) was significantly associated with DFS (p < 0.05) in PD-L1− patients with non-metastasis lung adenocarcinoma, not in PD-L1+ or lung squamous cell carcinoma patients. Moreover, stromal Siglec-15+ macrophages of Mφ-Siglec-15+PD-L1− patients were significantly more than those of Mφ-Siglec-15−PD-L1− patients (p = 0.002). We further found that Siglec-15+ macrophages polarized toward M2 and produced more IL-10, negatively associated with inflamed immunophenotype in PD-L1− patients and may inhibit CD8+T cells infiltration. In conclusion, PD-L1-independent Siglec-15+ macrophages contribute to the formation of an immunosuppressive microenvironment in non-metastasis lung adenocarcinoma patients, which may cause a higher risk of recurrence. Siglec-15 could be a potential target for normalizing cancer immunotherapy, benefiting patients who fail to respond to anti-PD-L1 therapy.
The oncofetal antigen Claudin 6 (CLDN6) is highly and specifically expressed in many solid tumors, and could be a promising treatment target. We report dose escalation results from the ongoing phase 1/2 BNT211-01 trial evaluating the safety and feasibility of chimeric antigen receptor (CAR) T cells targeting the CLDN6 with or without a CAR-T cell-amplifying RNA vaccine (CARVac) at two dose levels (DLs) in relapsed/refractory CLDN6-positive solid tumors. The primary endpoints were safety and tolerability, maximum tolerated dose and recommended phase 2 dose (RP2D). Secondary endpoints included objective response rate (ORR) and disease control rate. We observed manageable toxicity, with 10 out of 22 patients (46%) experiencing cytokine release syndrome including one grade 3 event and 1 out of 22 (5%) with grade 1 immune effector cell-associated neurotoxicity syndrome. Dose-limiting toxicities occurred in two patients at the higher DL, resolving without sequelae. CAR-T cell engraftment was robust, and the addition of CARVac was well tolerated. The unconfirmed ORR in 21 evaluable patients was 33% (7 of 21), including one complete response. The disease control rate was 67% (14 of 21), with stable disease in seven patients. Patients with germ cell tumors treated at the higher DL exhibited the highest response rate (ORR 57% (4 of 7)). The maximum tolerated dose and RP2D were not established as the trial has been amended to utilize an automated manufacturing process. A repeat of the dose escalation is ongoing and will identify a RP2D for pivotal trials. ClinicalTrials.gov Identifier: NCT04503278 . In the ongoing phase 1/2 BNT211-01 trial, CLDN6-specific chimeric antigen receptor (CAR)-T cells given with or without CARVac, a CAR-T cell-amplifying RNA vaccine, were well-tolerated and exhibited encouraging clinical activity in patients with relapsed or refractory CLDN6-positive solid tumors, with the highest response rate in patients with germ cell tumors.
WHAT IS ALREADY KNOWN ON THIS TOPICFocal adhesion kinase (FAK) kinase activity is elevated in human pancreatic ductal adenocarcinoma (PDAC) and FAK kinase inhibitors in combination with immunotherapy and chemotherapy can impair tumour growth in a mouse model of PDAC.FAK kinase inhibitors can...
The addition of immune checkpoint blockade to perioperative therapy was associated
with an increase in grade 3–4 treatment-related adverse events and adverse events
leading to treatment discontinuation. These findings provide safety insights for further
clinical trials assessing neoadjuvant or adjuvant immune checkpoint blockade therapy.
Clinicians should closely monitor patients for treatment-related adverse events to
prevent treatment discontinuations and morbidity from these therapies in earlier-stage
settings.
Although alterations in myeloid cells have been observed in COVID-19, the specific underlying mechanisms are not completely understood. Here, we examine the function of classical CD14+ monocytes in patients with mild and moderate COVID-19 during the acute phase of infection and in healthy individuals. Monocytes from COVID-19 patients display altered expression of cell surface receptors and a dysfunctional metabolic profile that distinguish them from healthy monocytes. Secondary pathogen sensing ex vivo leads to defects in pro-inflammatory cytokine and type-I IFN production in moderate COVID-19 cases, together with defects in glycolysis. COVID-19 monocytes switch their gene expression profile from canonical innate immune to pro-thrombotic signatures and are functionally pro-thrombotic, both at baseline and following ex vivo stimulation with SARS-CoV-2. Transcriptionally, COVID-19 monocytes are characterized by enrichment of pathways involved in hemostasis, immunothrombosis, platelet aggregation and other accessory pathways to platelet activation and clot formation. These results identify a potential mechanism by which monocyte dysfunction may contribute to COVID-19 pathology. Although myeloid cell dysfunction has been observed in COVID-19, the underlying mechanisms remain incompletely understood. Here, the authors demonstrate that monocytes from patients with mild to moderate COVID-19 show a blunted innate immune response and a pro-thrombotic signature following secondary SARS-CoV-2 challenge.
CD8+ T cells are end effectors of cancer immunity. Most forms of effective cancer immunotherapy involve CD8+ T cell effector function. Here, we review…
The therapeutic potential of interleukin (IL)-2 in cancer treatment has been known
for decades, yet its widespread adoption in clinical practice remains limited. Recently,
chimeric proteins of an anti-PD-1 antibody and suboptimal IL-2 variants were shown
to stimulate potent antitumor and antiviral immunity by inducing unique effector CD8+
T cells in mice. A similar subset of cytotoxic T cells is induced by depletion of
regulatory T cells (Tregs), suggesting IL-2 sequestration as a major mechanism through
which regulatory T cells suppress activated CD8+ T cells. Here, we present our view
of how IL-2-based biologicals can boost the antitumor response at a cellular level,
and propose that the role of Tregs following such treatments may have been previously
overestimated.
|
CAR T cell therapy has transformed the treatment of B cell cancers and is now being pursued by biotech companies for conditions as varied as systemic lupus erythematosus, diabetes and organ rejection.
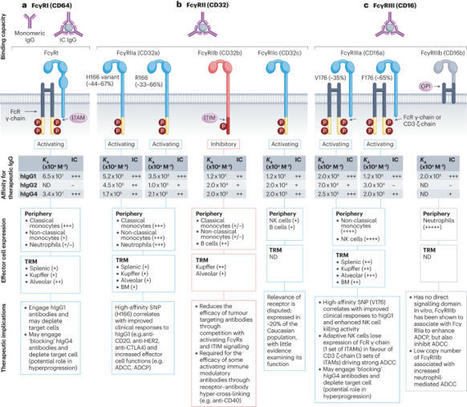
The discovery of both cytotoxic T lymphocyte-associated antigen 4 (CTLA4) and programmed cell death protein 1 (PD1) as negative regulators of antitumour immunity led to the development of numerous immunomodulatory antibodies as cancer treatments. Preclinical studies have demonstrated that the efficacy of immunoglobulin G (IgG)-based therapies depends not only on their ability to block or engage their targets but also on the antibody’s constant region (Fc) and its interactions with Fcγ receptors (FcγRs). Fc–FcγR interactions are essential for the activity of tumour-targeting antibodies, such as rituximab, trastuzumab and cetuximab, where the killing of tumour cells occurs at least in part due to these mechanisms. However, our understanding of these interactions in the context of immunomodulatory antibodies designed to boost antitumour immunity remains less explored. In this Review, we discuss our current understanding of the contribution of FcγRs to the in vivo activity of immunomodulatory antibodies and the challenges of translating results from preclinical models into the clinic. In addition, we review the impact of genetic variability of human FcγRs on the activity of therapeutic antibodies and how antibody engineering is being utilized to develop the next generation of cancer immunotherapies. Numerous immunomodulatory antibodies for cancer treatment have been developed following the discovery of negative regulators of antitumour immunity such as programmed cell death protein 1 (PD1) and cytotoxic T lymphocyte-associated antigen 4 (CTLA4). The efficacy of these antibodies is determined not only by their ability to block or engage their target but also by their interactions with Fcγ receptors (FcγRs). This Review outlines our current knowledge of these interactions and discusses how we can use this knowledge to generate more effective cancer immunotherapies in the future.
Abstract. Immunocompromised patients often fail to raise protective vaccine-induced immunity against the global emergence of severe acute respiratory syndrome c
Researchers have used a form of CRISPR, called base editing, to engineer T cells and hematopoietic stem cells to produce a potential “universal” CAR T-cell therapy for blood cancers.
On November 28th, the FDA issued a CAR-T warning regarding potential malignancies. What are the stakes for oncology?
Cyclin-dependent kinase (CDK) 4/6 inhibition in combination with endocrine therapy is the standard-of-care treatment for patients with advanced-stage hormone receptor-positive, HER2 non-amplified (HR+HER2−) breast cancer. These agents can also be administered as adjuvant therapy to patients with higher-risk early stage disease. Nonetheless, the clinical success of these agents has created several challenges, such as how to address acquired resistance, identifying which patients are most likely to benefit from therapy prior to treatment, and understanding the optimal timing of administration and sequencing of these agents. In this Review, we describe the rationale for targeting CDK4/6 in patients with breast cancer, including a summary of updated clinical evidence and how this should inform clinical practice. We also discuss ongoing research efforts that are attempting to address the various challenges created by the widespread implementation of these agents. Cyclin-dependent kinase (CDK) 4/6 inhibitors in combination with endocrine therapy have become the standard-of-care therapy for patients with advanced-stage hormone receptor-positive breast cancer. However, this success has created several challenges, such as the need to better understand resistance to these agents and develop novel therapies accordingly. Here, the authors provide an update on the clinical activity of the established CDK4/6 inhibitors along with a summary of ongoing research efforts attempting to address the new challenges created by the success of these agents.
Results of this prospective cohort study suggest an association between antidrug antibodies and nonresponse to bDMARDs in patients with RA. Monitoring antidrug antibodies could be considered in the treatment of these patients, particularly nonresponders to biologic RA drugs.
A combination of immune stimulants and immunotherapy that transforms tumors into cancer vaccine-producing factories has shown remarkable promise in a phase 1/2 trial, although only in 30% of patients.
Pritzker Molecular Engineering researchers led by Prof. Jeffrey Hubbell showed that their compound can eliminate the autoimmune reaction associated with multiple sclerosis in a laboratory setting.
Chimeric antigen receptor (CAR) T cells have been approved for use in patients with B cell malignancies or relapsed and/or refractory multiple myeloma, yet efficacy against most solid tumours remains elusive. The limited imaging and biopsy data from clinical trials in this setting continues to hinder understanding, necessitating a reliance on imperfect preclinical models. In this Perspective, I re-evaluate current data and suggest potential pathways towards greater success, drawing lessons from the few successful trials testing CAR T cells in patients with solid tumours and the clinical experience with tumour-infiltrating lymphocytes. The most promising approaches include the use of pluripotent stem cells, co-targeting multiple mechanisms of immune evasion, employing multiple co-stimulatory domains, and CAR ligand-targeting vaccines. An alternative strategy focused on administering multiple doses of short-lived CAR T cells in an attempt to pre-empt exhaustion and maintain a functional effector pool should also be considered. Despite some success in patients with certain B cell malignancies and relapsed and/or refractory multiple myeloma, studies testing chimeric antigen receptor (CAR) T cells in patients with advanced-stage solid tumours have been largely unsuccessful, with a few notable exceptions. In this Perspective, the author provides some possible reasons for the failures of most CAR T cell-based approaches and suggests strategies that might address some of these challenges.
|