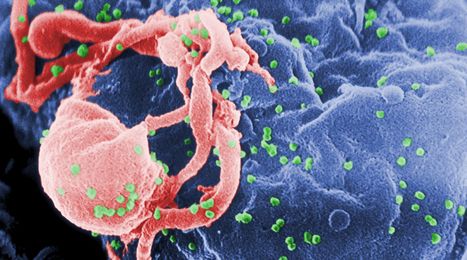
Lymphocyte receptor diversity is generated by recombining variable, diversity, and joining (VDJ) gene segments of the immunoglobulin and T-cell receptor (TCR) loci. V(D)J recombination requires DNA breakage, a process mediated by recombination-activating gene (RAG) 1 and 2. RAG deficiency was initially described in patients with the T−B− severe combined immunodeficiency (SCID) phenotype1; however, the spectrum of the disease has expanded to include Omenn syndrome, cytomegalovirus infection with γδ T-cell expansion, combined immunodeficiency with granuloma, and isolated CD4+ lymphopenia.2, 3, 4, 5, 6 The pleomorphic manifestations of RAG deficiency are partially explained by residual RAG activity, with null mutations producing an SCID phenotype and hypomorphic mutations presenting more variably.2, 7 Although autoimmunity is a known feature of aberrant RAG function, it has never been described as the primary manifestation of the disease in an infant. We describe a novel presentation of RAG deficiency characterized by the presence of B cells and early-onset autoimmunity.


 Your new post is loading...
Your new post is loading...


















X-linked agammaglobulinemia (XLA) is an inherited immunodeficiency that results from mutations within the gene encoding Bruton’s tyrosine kinase (BTK). Many XLA-associated mutations affect splicing of BTK pre-mRNA and severely impair B cell development. Here, we assessed the potential of antisense, splice-correcting oligonucleotides (SCOs) targeting mutated BTK transcripts for treating XLA. Both the SCO structural design and chemical properties were optimized using 2′-O-methyl, locked nucleic acid, or phosphorodiamidate morpholino backbones. In order to have access to an animal model of XLA, we engineered a transgenic mouse that harbors a BAC with an authentic, mutated, splice-defective human BTK gene. BTK transgenic mice were bred onto a Btk knockout background to avoid interference of the orthologous mouse protein. Using this model, we determined that BTK-specific SCOs are able to correct aberrantly spliced BTK in B lymphocytes, including pro–B cells. Correction of BTK mRNA restored expression of functional protein, as shown both by enhanced lymphocyte survival and reestablished BTK activation upon B cell receptor stimulation. Furthermore, SCO treatment corrected splicing and restored BTK expression in primary cells from patients with XLA. Together, our data demonstrate that SCOs can restore BTK function and that BTK-targeting SCOs have potential as personalized medicine in patients with XLA.
GO TO THE JOURNAL OF CLINICAL INVESTIGATION MANUSCRIPT:
http://www.jci.org/articles/view/76175